Trình tự DNA của bạn có thể được sử dụng khá dễ dàng với phần mềm Blast và Mega – các chương trình này có thể giúp xây dựng cây phát sinh. Bạn có thể đặt câu hỏi như mối quan hệ tiến hóa giữa một tập hợp các chuỗi từ các loài khác nhau là gì? Hoặc có những dòng vi khuẩn nào xuất hiện?
BLAST
Như bất kỳ nhà sinh học học nào cũng có thể biết, bước đầu tiên của bạn đối với một chuỗi trình tự mới sẽ là sử dụng BLAST, công cụ Basic Local Alignment Search. Công cụ tiện lợi nhưng mạnh mẽ này sẽ lắp ghép trình tự của bạn với hàng triệu chuỗi được lưu trữ trong các cơ sở dữ liệu genomic và nucleotide. Công cụ này cho kết quả là các trình tự tương tự nhất với trình tự của bạn. Nó cũng cung cấp những hiểu biết chuyên sâu để có thể nhận dạng của các chuỗi đó. Kết quả bao gồm sự tương đồng giữa các loài và các mô tương tự nhau. Công cụ Blast rất quan trọng vì nó giúp xác nhận rằng các trình tự tương đồng và không phải là liên kết ngẫu nhiên.
Những điều cơ bản của việc sử dụng BLAST để tìm kiếm trình tự nucleotide sẽ được đề cập đến trong bài viết này. Dưới đây là một vài giới thiệu ngắn gọn có liên quan đến BLAST được tìm thấy trên trang web của NCBI:
- BLASTN: So sánh trình tự nucleotide của bạn với các trình tự nucleotide trong GenBank, kho của NCBI sẽ cho các trình tự nucleotide.
- BLASTX: So sánh sáu khung dịch mã khác nhau (mở khung đọc) của trình tự nucleotide của bạn tới chuỗi amino axit trong cơ sở dữ liệu Protein của NCBI. Đây là một cách tuyệt vời để tìm ra các sản phẩm và chức năng có thể có của chuỗi của bạn!
- MegaBLAST: So sánh trình tự của bạn với các trình tự nucleotide khác, tối ưu hóa để tìm các chuỗi tương tự nhau của các loài liên quan, điều này làm nên một mạng lưới chặt chẽ hơn.
Sắp xếp nhiều trình tự (MSA)
Nhiều sự tương đồng được phát hiện qua Blast có thể được liên kết bằng cách sử dụng các thuật toán như ClustalW hoặc MUSCLE. Tôi thích sử dụng MEGA (Molecular Evolutionary Genetics Analysis) vì nó chứa đựng rất nhiều chức năng.
Để bắt đầu sắp xếp các chuỗi của bạn, khởi chạy Alignment Explorer bằng cách chọn Align /Edit/Build Alignment. Nó nằm trên thanh khởi động của cửa sổ chính MEGA. Từ Menu chính của Alignment Explorer, chuyển đến Web-> Query GenBank. Điều này cho phép bạn thêm từng chuỗi một cho các liên kết của bạn vào visual explorer. Sau khi thêm tất cả các chuỗi, bạn tùy chọn để sắp xếp chúng bằng cách sử dụng một đến hai chương trình khác nhau thường được sử dụng – bạn có thể sử dụng phần mềm ClustalW hoặc MUSCLE.
Chọn ClustalW hoặc MUSCLE?
Hai chương trình có sự khác nhau trong cách hoạt động. ClustalW sử dụng một thuật toán tiến bộ cho sự liên kết. Nó sắp xếp hai dãy tại mỗi bước, sau đó sắp xếp liên kết với một dãy khác, v.v … MUSCLE là viết tắt của Multiple Sequence Comparison by Log-Expectation. Nó đạt được kết quả tốt hơn ClustalW qua các thông số quan trọng, các thông số này bao gồm độ chính xác của liên kết cũng như sự phức tạp về thời gian và không gian thấp hơn bằng cách sử dụng các liên kết tịnh tiến chứ không phải lặp đi lặp lại.
Vào Alignment, và chọn Align by Muscle. Với người dùng ban đầu, các giá trị cài sẵn có thể sử dụng, vì chúng phục vụ mục đích của hầu hết mọi người. Đầu ra của bạn nên để giống như hình ảnh dưới đây:
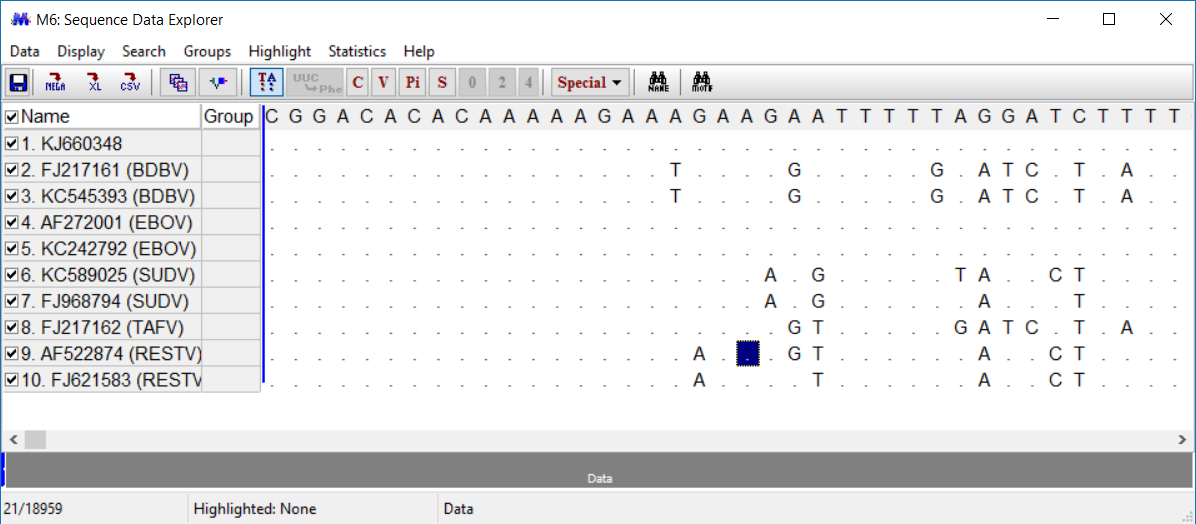
Lưu sắp xếp của bạn dưới dạng tệp .meg. Bằng cách này, bạn có thể sử dụng nó sau mà không cần phải dành nhiều thời gian thêm và sắp xếp các trình tự một lần nữa.
Xây dựng cây phân loại với MEGA Phiên bản 6
Bây giờ đến phần thú vị! MEGA có rất nhiều lựa chọn để xây dựng cây phát sinh, bao gồm cây UPGMA, Maximum Parsimony, Neighbor-Joining, và Maximum Likelihood. Đây là những cách tiếp cận khác nhau để xây dựng cây, mỗi lựa chọn có ưu và nhược điểm riêng, và phù hợp cho mục đích cụ thể của bạn. Đối với một phương pháp đã chọn, Mega sẽ giúp bạn tìm ra mô hình tốt nhất cho tỷ lệ thay thế trình tự DNA hoặc protein của bạn.
Để xây dựng một cây phát sinh, đóng các liên kết tìm kiếm và trở lại cửa sổ chính MEGA. Chúng ta sẽ xây dựng một cây kết Neighbor-Joining để xem nhanh các trình tự và quan hệ của chúng với nhau. Bạn luôn có thể quay lại và vẽ lại cây bằng các phương pháp khác!
Chọn Phylogeny- Construct/Test Neighbor-Joining Tree, và chọn lưu tập tin với đuôi .meg từ Alignment Explorer trong hộp thoại mở. Sau khi chọn và nhấp vào Computer, bạn sẽ nhận được một cây giống như sau:
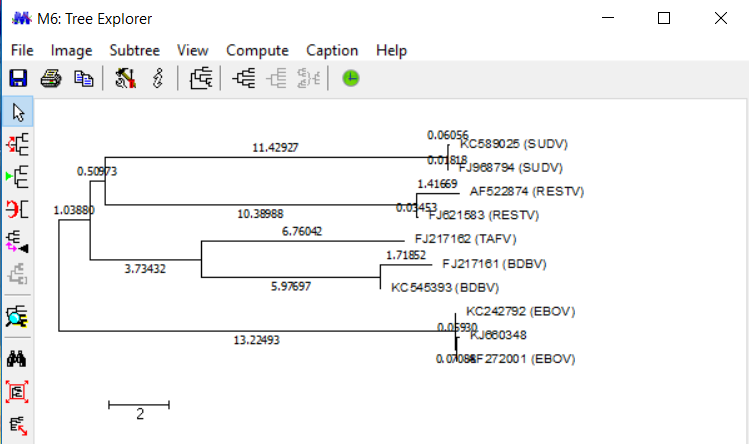
Để dễ đọc hơn hãy nhấp vào nút ở trên (Display Only Topology) (hình ảnh bên dưới).
Tree Explorer
Cây này cho chúng ta rất nhiều thông tin về trình tự. Giờ đây rõ ràng là trình tự Zaire Ebolavirus từ Gueckedou ở Guinea, giống nhất với dòng Mayinga (chuỗi AF272001.1). Cả hai giống này gần giống nhất với dòng của Gabon, tương tự như của Tai Forest hay Sudan. Đây là một thực tế đáng ngạc nhiên khi xem xét vị trí địa lý của chúng ở Châu Phi. Guinea nằm ở Bắc Phi và Gabon nằm qua Vịnh Guinea. Điều này cho thấy rằng dơi có thể là vật lây lan chủ yếu virus ebola giữa các vị trí này.
BLAST và MEGA sẽ giúp bạn bắt đầu phân tích bộ gen và hiểu được trình tự dữ liệu. Đây là một giới thiệu rất ngắn về sức mạnh của MEGA. Lưu ý, độ tin cậy của cây có thể được ước lượng bằng cách sử dụng phương pháp bootstrap.
Tham khảo
1. Stecher, G., Liu, L., Sanderford, M., Peterson, D., Tamura, K., & Kumar, S. MEGA-MD: molecular evolutionary genetics analysis software with mutational diagnosis of amino acid variation. Bioinformatics 30, no. 9 (2014): doi:10.1093/bioinformatics/btu018.
Nguồn gốc bài viết: Bitesizebio
Người dịch: Trà My
Biên tập: Sinhhocvietnam.com
