Nguyễn Quang Hưng
Senior Member
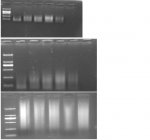
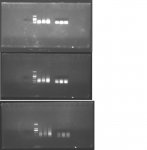
Chào các bạn trong diễn đàn, mình có gặp một vấn đề trong quá trình làm thí nghiệm mong các bạn giải thích giúp nhé. Mình dùng Agarose 1.1% điện di DNA trong khoảng 15 phút, kiểm tra kết quả thấy vệt sáng ở vạch 100 hoặc 200 bp, nhưng nếu điện di tiếp khoảng 10 phút nữa thì vệt sáng đó lại chạy dài theo hướng ngược lên trên là sao vậy ?.  Thanks !
Thanks !
")